Example 5: Example of torsional scan using a norfloxacin analog¶
Torsional scan recipe¶
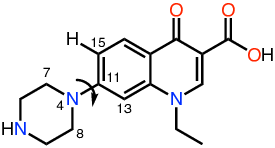
In this example we are going to use ParaMol’s torsional scan Task (ParaMol.Tasks.torsions_scan.TorsionScan) to parametrize a torsion of the norfloxacin analog represented above.
First of all, we are going to symmetrize the ParaMol Force Field so that it respects atom-type symmetries and write it to a file in order to choose what torsions we want to parametrize.
# ParaMol imports
from ParaMol.System.system import *
from ParaMol.MM_engines.openmm import *
from ParaMol.Utils.Symmetrizers.amber_symmetrizer import *
# --------------------------------------------------------- #
# Preparation #
# --------------------------------------------------------- #
# Create the OpenMM engine for norfloxacin
openmm_system = OpenMMEngine(init_openmm=True, topology_format='AMBER', top_file='norfloxacin.prmtop', crd_format='AMBER', crd_file='norfloxacin.inpcrd')
# Create ParaMol System
norfloxacin = ParaMolSystem(name="norfloxacin", engine=openmm_system, n_atoms=41)
# Create ParaMol's force field representation
norfloxacin.force_field.create_force_field()
# --------------------------------------------------------- #
# Symmetrize ParaMol ForceField #
# --------------------------------------------------------- #
# Symmetry ParaMol ForceField so that it respects atom-type symmetries
amber_symmetrizer = AmberSymmetrizer(top_file="norfloxacin.prmtop")
amber_symmetrizer.get_symmetries()
amber_symmetrizer.symmetrize_force_field(norfloxacin.force_field)
# Write symmetrized Force-Field to file
norfloxacin.force_field.write_ff_file("norfloxacin_symm.ff")
We are interested in parametrizing the torsions around the C11-N4 bond. Hence, the next step is to modify the ParaMol Force Field file in order to set as optimizable all torsions’s parameters involved in the rotation of C11 and N4, specifically the phase and barrier heights of these torsions (ParaMol cannot optimize the periodicity). This can be achieved by performing the following modifications on the ParaMol Force Field file:
87 7 4 11 13 2.00000000 3.14159400 4.39320000 0 0 0 T8
88 7 4 11 15 2.00000000 3.14159400 4.39320000 0 0 0 T8
...
92 8 4 11 13 2.00000000 3.14159400 4.39320000 0 0 0 T8
93 8 4 11 15 2.00000000 3.14159400 4.39320000 0 0 0 T8
87 7 4 11 13 2.00000000 3.14159400 4.39320000 0 1 1 T8
88 7 4 11 15 2.00000000 3.14159400 4.39320000 0 1 1 T8
...
92 8 4 11 13 2.00000000 3.14159400 4.39320000 0 1 1 T8
93 8 4 11 15 2.00000000 3.14159400 4.39320000 0 1 1 T8
Now that we have done the necessary changes in the ParaMol Force Field file, we are ready to perform the torsional scan and subsequent parameters’s optimization. Luckily, as all the torsions around the C11-N4 are of the same type and, therefore, they share the same set of parameters, we only need to perform the torsional scan of one of the torsions with symmetry T8. Furthermore, when asking ParaMol to create its Force Field representation, we need to provide the modified ParaMol Force Field file so that ParaMol creates its internal representation of the Force Field from this file. The same procedure can be done to set special constraints or to optimize other parameters.
# ParaMol imports
from ParaMol.System.system import *
from ParaMol.MM_engines.openmm import *
from ParaMol.QM_engines.qm_engine import *
# ParaMol Tasks imports
from ParaMol.Tasks.parametrization import *
from ParaMol.Tasks.torsions_scan import *
from ParaMol.Utils.settings import *
from ParaMol.Utils.Symmetrizers.amber_symmetrizer import *
# --------------------------------------------------------- #
# Preparation #
# --------------------------------------------------------- #
# Create the OpenMM engine for norfloxacin
openmm_system = OpenMMEngine(init_openmm=True, topology_format='AMBER', top_file='norfloxacin.prmtop', crd_format='AMBER', crd_file='norfloxacin.inpcrd')
# Create ParaMol System
norfloxacin = ParaMolSystem(name="norfloxacin", engine=openmm_system, n_atoms=41)
# Create ParaMol's force field representation
norfloxacin.force_field.create_force_field(ff_file="norfloxacin_symm.ff")
# Create ParaMol settings instance
paramol_settings = Settings()
# The objective function will contain an energy and a regularization term
paramol_settings.properties["include_energies"] = True
paramol_settings.properties["include_forces"] = False # One should not include forces when a torsional scan
paramol_settings.properties["include_regularization"] = True
# --------------------------------------------------------- #
# Symmetrize ParaMol ForceField #
# --------------------------------------------------------- #
# Symmetry ParaMol ForceField so that it respects atom-type symmetries
amber_symmetrizer = AmberSymmetrizer(top_file="norfloxacin.prmtop")
amber_symmetrizer.get_symmetries()
# Do not set force field to amber_format as we are already reading it with the correct symmetries from the "norfloxacin_symm.ff"
# amber_symmetrizer.set_force_field_to_amber_format(norfloxacin.force_field)
# --------------------------------------------------------- #
# Set the QM Engine #
# --------------------------------------------------------- #
# Create the ASE calculator
from ase.calculators.dftb import *
calc = Dftb(Hamiltonian_='DFTB', # line is included by default
Hamiltonian_MaxAngularMomentum_='',
Hamiltonian_MaxAngularMomentum_H='s',
Hamiltonian_MaxAngularMomentum_O='p',
Hamiltonian_MaxAngularMomentum_C='p',
Hamiltonian_MaxAngularMomentum_N="p",
Hamiltonian_Dispersion="DftD3 { \n s6=1.000 \n s8=0.5883 \n Damping = BeckeJohnson { \n a1=0.5719 \n a2=3.6017 \n } \n }",
Hamiltonian_SCC='Yes',
Hamiltonian_SCCTolerance=1e-8, )
# Alternative, we could set the calculator in the settings
paramol_settings.qm_engine["ase"]["calculator"] = calc
# --------------------------------------------------------- #
# Perform the Torsional Scan #
# --------------------------------------------------------- #
torsion_to_scan = [[7, 4, 11, 15]]
scan_settings = [[-180.0, 180.0, 90.0]]
torsion_scan = TorsionScan()
torsion_scan.run_task(paramol_settings, [norfloxacin], torsion_to_scan, scan_settings, optimize_qm_before_scan=True)
# Save coordinates and energies into .nc file
norfloxacin.write_data("norfloxacin_scan.nc")
# --------------------------------------------------------- #
# Parametrize the Torsion #
# --------------------------------------------------------- #
parametrization = Parametrization()
systems, parameter_space, objective_function, optimizer = parametrization.run_task(paramol_settings, [norfloxacin])
# Update AMBER symmetrizer with new parameters
amber_symmetrizer.update_term_types_parameters(parameter_space.optimizable_parameters)
# Write AMBER topology file (.prmtop) and .frcmod file
amber_symmetrizer.save("norfloxacin_opt.prmtop")
amber_symmetrizer.save_frcmod("norfloxacin_opt.frcmod")
Alternative method (preferred)¶
Alternatively, we could have avoided changing manually the Force Field file if we had used the ParaMol.Force_field.force_field.ForceField.optimize_torsions_by_symmetry function. Hence, the whole procedure could be performed using the following code:
# ParaMol imports
from ParaMol.System.system import *
from ParaMol.MM_engines.openmm import *
from ParaMol.QM_engines.qm_engine import *
# ParaMol Tasks imports
from ParaMol.Tasks.parametrization import *
from ParaMol.Tasks.torsions_scan import *
from ParaMol.Utils.settings import *
from ParaMol.Utils.Symmetrizers.amber_symmetrizer import *
# --------------------------------------------------------- #
# Preparation #
# --------------------------------------------------------- #
# Create the OpenMM engine for norfloxacin
openmm_system = OpenMMEngine(init_openmm=True, topology_format='AMBER', top_file='norfloxacin.prmtop', crd_format='AMBER', crd_file='norfloxacin.inpcrd')
# Create ParaMol System
norfloxacin = ParaMolSystem(name="norfloxacin", engine=openmm_system, n_atoms=41)
# Create ParaMol's force field representation
norfloxacin.force_field.create_force_field()
# Create ParaMol settings instance
paramol_settings = Settings()
# The objective function will contain an energy and a regularization term
paramol_settings.properties["include_energies"] = True
paramol_settings.properties["include_forces"] = False # One should not include forces when a torsional scan
paramol_settings.properties["include_regularization"] = True
# --------------------------------------------------------- #
# Symmetrize ParaMol ForceField #
# --------------------------------------------------------- #
# Symmetry ParaMol ForceField so that it respects atom-type symmetries
amber_symmetrizer = AmberSymmetrizer(top_file="norfloxacin.prmtop")
amber_symmetrizer.get_symmetries()
amber_symmetrizer.symmetrize_force_field(norfloxacin.force_field)
# --------------------------------------------------------- #
# Set the QM Engine #
# --------------------------------------------------------- #
# Create the ASE calculator
from ase.calculators.dftb import *
calc = Dftb(Hamiltonian_='DFTB', # line is included by default
Hamiltonian_MaxAngularMomentum_='',
Hamiltonian_MaxAngularMomentum_H='s',
Hamiltonian_MaxAngularMomentum_O='p',
Hamiltonian_MaxAngularMomentum_C='p',
Hamiltonian_MaxAngularMomentum_N="p",
Hamiltonian_Dispersion="DftD3 { \n s6=1.000 \n s8=0.5883 \n Damping = BeckeJohnson { \n a1=0.5719 \n a2=3.6017 \n } \n }",
Hamiltonian_SCC='Yes',
Hamiltonian_SCCTolerance=1e-8, )
# Alternative, we could set the calculator in the settings
paramol_settings.qm_engine["ase"]["calculator"] = calc
# --------------------------------------------------------- #
# Perform the Torsional Scan #
# --------------------------------------------------------- #
torsion_to_scan = [[7, 4, 11, 15]]
scan_settings = [[-180.0, 180.0, 90.0]]
torsion_scan = TorsionScan()
torsion_scan.run_task(paramol_settings, [norfloxacin], torsion_to_scan, scan_settings, optimize_qm_before_scan=True)
# Save coordinates and energies into .nc file
norfloxacin.write_data("norfloxacin_scan.nc")
# --------------------------------------------------------- #
# Parametrize the Torsion #
# --------------------------------------------------------- #
norfloxacin.force_field.optimize_torsions_by_symmetry(torsion_to_scan)
norfloxacin.force_field.write_ff_file("norfloxacin_symm.ff")
parametrization = Parametrization()
systems, parameter_space, objective_function, optimizer = parametrization.run_task(paramol_settings, [norfloxacin])
# Update AMBER symmetrizer with new parameters
amber_symmetrizer.update_term_types_parameters(parameter_space.optimizable_parameters)
# Write AMBER topology file (.prmtop) and .frcmod file
amber_symmetrizer.save("norfloxacin_opt.prmtop")
amber_symmetrizer.save_frcmod("norfloxacin_opt.frcmod")